Cyclic peptides and cyclic proteins are useful tools in chemical biology and pharmaceutical science. [1] Their constrained conformation often results in increased resistance to proteolysis and higher receptor-binding affinities as compared to their linear counterparts. The classical methods for the synthesis of amide-cyclized peptides rely on protected linear precursors which are lactamized in organic solvents, either at high dilution or using pseudo-dilution on a solid support. [2] A major limitation of these methods is that the acylating moiety has to be highly enthalpically activated during the lactamization process. Epimerization may take place [3] and a high dilution (typically at submillimolar concentrations) is required to prevent polymeri- zation. Furthermore, protected peptides are often difficult to dissolve as their size increases. Therefore, application of the classical methods to the cycliza- tion of relatively large peptides remains very difficult.
To overcome the above problems, Tam, [4] Muir, [5] and others [6] developed alternative methods to cyclize fully unpro- tected peptides in aqueous solution. These methods usually in- volve the native chemical ligation (NCL) [7] of a C-terminal thio- ester with an N-terminal Cys. Besides NCL, the use of thiazolidine-forming ligation [8] and traceless Staudinger ligation [9] in peptide cyclization has also been tested. [10] A key advantage of these methods is that the cyclic peptides are synthesized in an epimerization-free manner. Moreover, these methods produce few polymerized byproducts, so that the cyclization may be conducted at moderately high concentrations. These advantages are especially useful for the the end-to-end cyclization of large peptides that are rich in Cys residues. Nonetheless, cyclic peptides without any Cys can also be synthesized by using modified versions of the methods. [6,11]
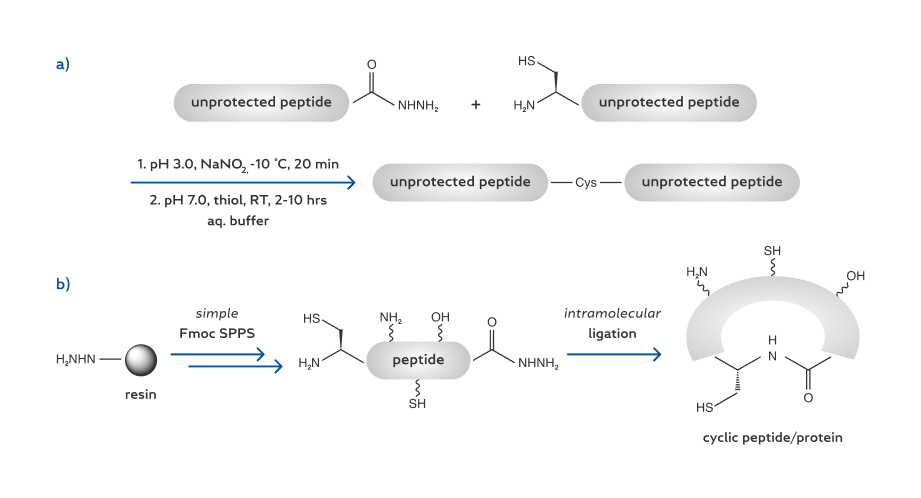
Very recently we reported a new ligation between a C-terminal peptide hydrazide and an N-terminal Cys under the activation of NaNO (Scheme 1). [12] This ligationis, inessence, a modified version of NCL with an in situ conversion of a peptide hydrazide to a thioester. One of its important advantages is that peptide hydrazides can be easily prepared through routine Fmoc solid-phase peptide synthesis (SPPS). This feature compares favorably with the methods previously used to generate thioesters for peptide cyclization (e.g. Boc SPPS, [13] use of “safety-catch” linkers, [14] and post-SPPS thioesterification) [15].
Thus we were inspired to develop a new peptide cyclization method on the basis of the hydrazide ligation (Scheme 1). Our study showed that this new method provides an easier and less expensive means to prepare various cyclic peptides and cyclic proteins. The synthesis of cyclic peptides also extends the concept and utility of the ligation of peptide hydrazides.
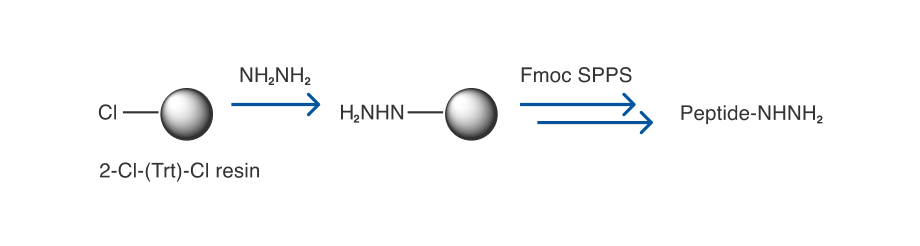
Our study began with the conversion of a model peptide 1a to a cyclic peptide 2a, which is an analogue of the toxin a-amanitin produced by Amanite mushrooms (Table1) [16]. The linear precursor 1a was synthesized by using the commercially available 2-Cl-(Trt)-Cl resin (Scheme 2) [17]. A hydrazine-Trt(2-Cl) resin was readily prepared as the key intermediate, and the subsequent steps were standard Fmoc SPPS. It is important to note that synthesis with the hydrazine-Trt(2-Cl) resin tends to produce peptide hydrazides with higher yields and improved purities as compared to our previous method with the Wang resin. [12]
The cyclization of 1a involves two steps that are carried out in a one-pot fashion. In the first step, 1a was treated with 10 equiv of NaNO in an aqueous phosphate buffer at pH 3.0 and -10 ˚ C. After 20 min, a thiol additive was added and the pH was adjusted to 7.0 to initiate the second step. The second step was allowed to proceed for 30 min at RT before the yield was determined by HPLC. As shown in Table 1, various thiol additives were used for the cyclization, among which 4-mer-captophenylacetic acid (MPAA) [18] shows the optimal perfor- mance (entries 1–4). Although 10 equiv of MPAA additive is already sufficient for the reaction (entries 5–7), the use of 40 equiv of MPAA provides a higher yield (92.9 %). The aque- ous phosphate buffer containing 6.0m guanidinium chloride was a good solvent system for cyclization. Nonetheless, a mix- ture of an organic solvent with aqueous phosphate buffer can also be used as the medium for the transformation (entries 8– 11). Furthermore, the cyclization step is not sensitive to the pH value (entries 12–13). Good yields can be obtained for moder- ately high concentrations (e.g. 10 mm) of the reactant (entries 14–15).
To test whether or not the cyclization caused epimerization at the C-terminal amino acid, we compared the cyclization of 1 a and H-Cys-Asn-Pro-Ile-Trp-Gly-Ile-(D)Phe-NHNH (1 b). The results showed that the extent of racemization was lower than 1% (Supporting Information). To understand the mechanism of the cyclization reaction, we analyzed the intermediates of the cyclization of 1 c (Figure 1). It was found that NaNO can clean- ly convert 1 c to a peptide azide 1 c’ in 20 min at pH 3.0. After the addition of MPAA and pH adjustment to 7, 1 c’ immediately transforms into a peptide thioester 1c’’, which then cyclizes presumably through NCL to give the desired cyclic peptide 2c.
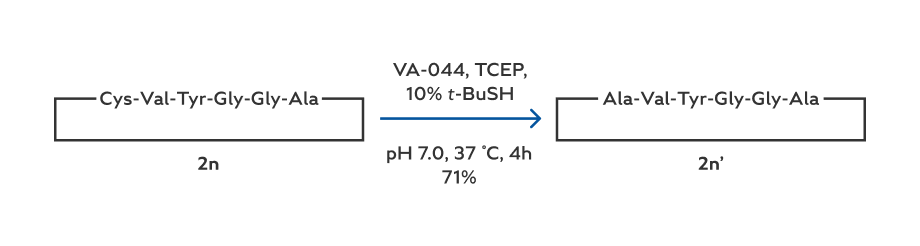
In addition to cyclooctapeptides, the hydrazide-based method can also be used to prepare smaller cyclic peptides. This was demonstrated by the synthesis of cyclopentapeptides 2 m and cyclohexapeptides 2 n’ (Stellaria cyclopeptide, [19] Scheme 3). Furthermore, as for the cyclohexapeptide 2n’, its Ala-to-Cys mutant 2 n was readily prepared in 54% isolated yield based on the initial resin loading (entry 12, Table 2). The target compound 2 n’ was then obtained in 71 % yield by using the well-established method of desulfurization.[20] Note that in addition to Ala, the cyclization/desulfurization strategy has also been shown to be possible with other N-terminal amino acids including Phe, Val, Leu, Lys, Thr, and Pro.[11] This means that the hydrazide-based method can be used to pre- pare various cyclic peptides that do not contain any Cys resi- due. Furthermore, the hydrazide-based method is also compat- ible with the acetamidomethyl (Acm) group[21] when the inter- nal Cys requires protection in a peptide hydrazide (Supporting Information).
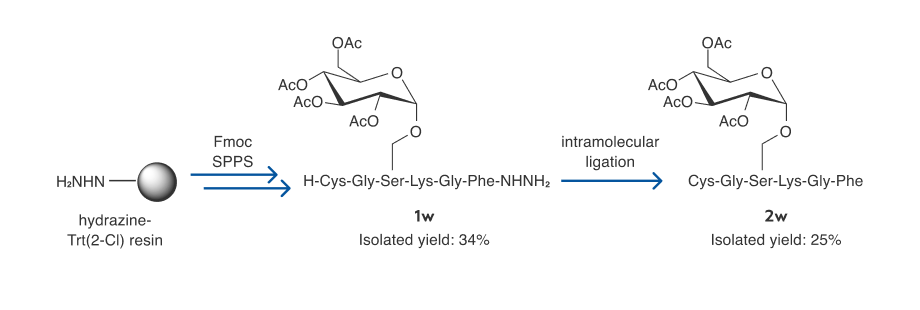
Further tests on various longer cyclic peptides (9–42 mers) show that these target compounds can be prepared in decent isolated yields (18–52%) with the hydrazide-based method (entries 13–20). Finally, a glycosylated cyclic peptide [22] 2 w can be readily prepared in 25% isolated yield (based on the initial resin loading) through the Fmoc SPPS and hydrazide-based intramolecular ligation (Scheme 4). This particular application is an important advant- age of the present approach as compared to the Boc SPPS method for thioester synthesis—cyclic peptides or proteins with delicate modifications can be more readily synthesized using the present method.
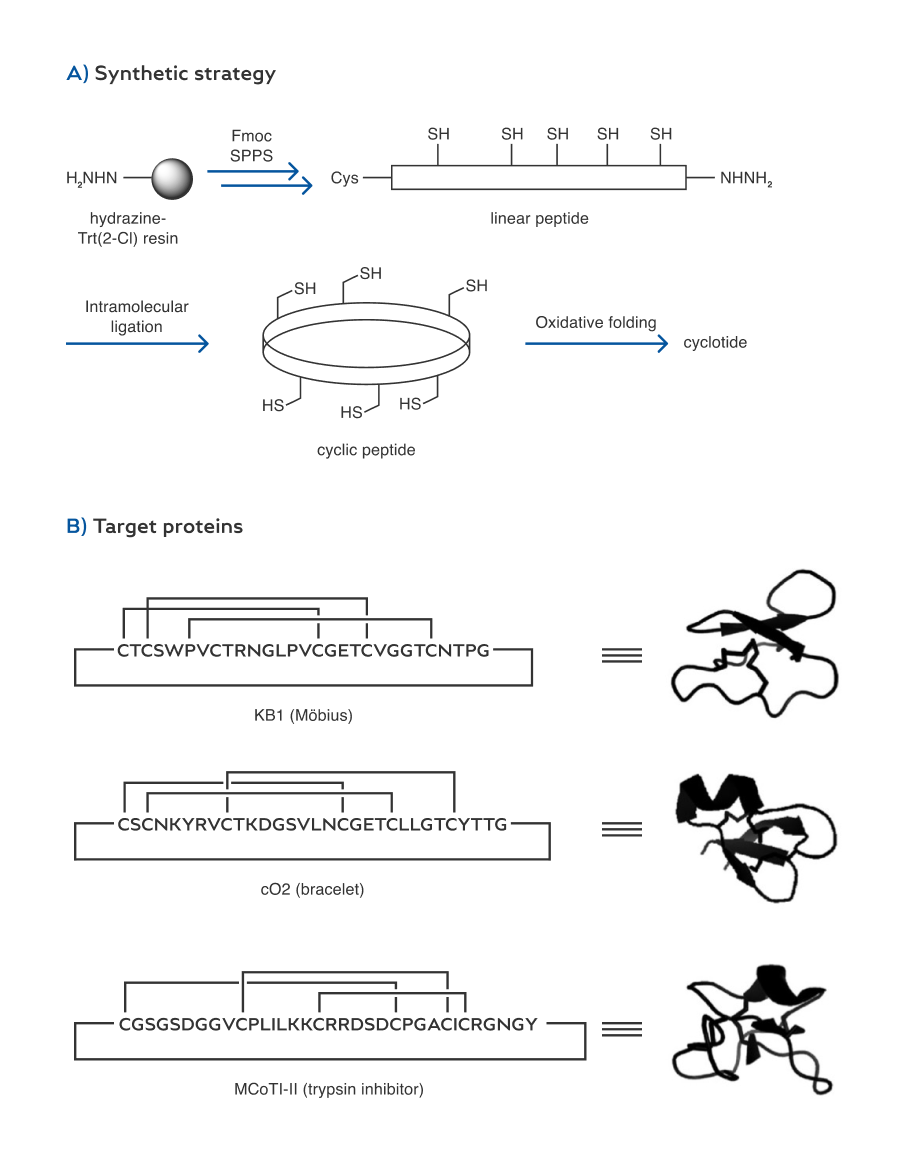
To test the application of the hydrazide-based method to the preparation of cyclic proteins, we chose cyclotides as syn- thetic targets. Cyclotides are backbone-cyclized mini-proteins isolated from several plant families.[23] They typically contain ca. 30 amino acids with a knotted arrangement of three disul- fide bonds. This unique structure provides exceptional stability to chemical, enzymatic and thermal treatments. As a result, cyclotides have been implicated as an ideal scaffold for the development of stable protein-based drugs.[24] In this context, a number of groups have studied the chemical synthesis of cyclotides.[4b,c,5b,25] The ability to prepare various cyclotide derivatives at low cost is critical to their structure-activity relationship studies and therapeutic applications.
Some previous syntheses of cyclotides relied on the use of Boc SPPS to generate the peptide thioester precursors for subsequent intramolecular ligation.[4,24,25] Nonetheless, in many studies on cyclotides the Fmoc SPPS method was used due to various operational reasons. Various approaches such as the use of “safety-catch” linkers and post-SPPS thioester synthesis have been tested in order to prepare the peptide thioesters.[25] Here we showed that the three subfamilies (i.e. Mçbius, brace- let, and trypsin inhibitor) of cyclotides can all be readily synthesized with high efficiencies by using the hydrazide-based ligation method.


| Cyclic peptide | Structure | Isolated yield of linear precursor [%][a] | Isolated yield of cyclic peptide [%][a] | |
|---|---|---|---|---|
| 1 | 2 c (8-mer) |

| 76 | 59 |
| 2 | 2 d (8-mer) |

| 65 | 55 |
| 3 | 2 e (8-mer) |

| 72 | 63 |
| 4 | 2 f (8-mer) |

| 70 | 60 |
| 5 | 2 g (8-mer) |

| 72 | 57 |
| 6 | 2 h (8-mer) |

| 55 | 42 |
| 7 | 2 i (8-mer) |

| 75 | 65 |
| 8 | 2 j (8-mer) |

| 60 | 45 |
| 9 | 2 k (8-mer) |

| 59 | 39 |
| 10 | 2 l (8-mer) |

| 46 | 35 |
| 11 | 2 m (5-mer) |

| 72 | 48 |
| 12 | 2 n (6-mer) |

| 77 | 54 |
| 13 | 2 o (8-mer) |

| 58 | 45 |
| 14 | 2 p (8-mer) |

| 70 | 52 |
| 15 | 2 q (9-mer) |

| 68 | 48 |
| 16 | 2 r (11-mer) |

| 38 | 30 |
| 17 | 2 s (13-mer) |

| 53 | 45 |
| 18 | 2 t (16-mer) |

| 45 | 34 |
| 19 | 2 u (18-mer) |

| 52 | 41 |
| 20 | 2 v (42-mer) |

| 23 | 18 | [a] Isolated yields were calculated based upon the weight of each peptide produced after lyophilization and loading of the first amino acid on the resin. |
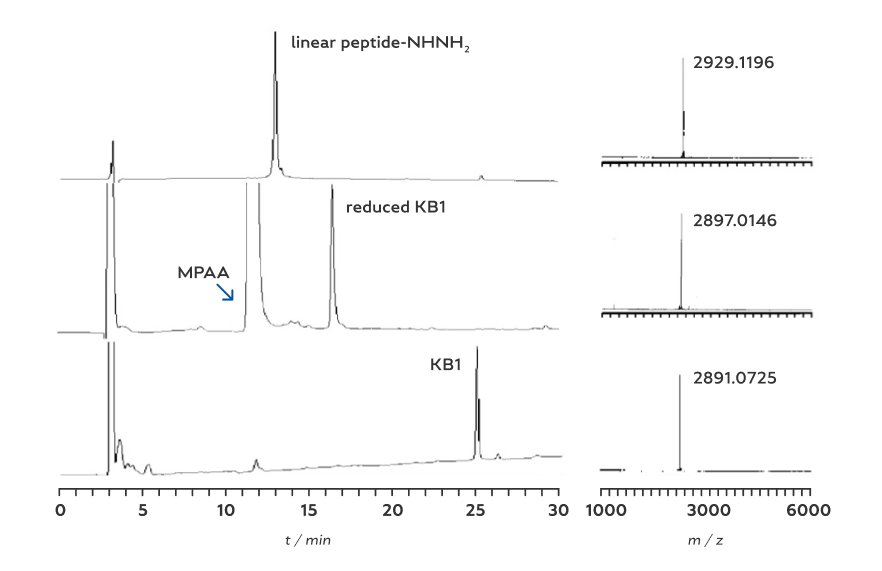
Our approach to the synthesis of cyclotides involves three distinct stages: synthesis of linear peptide hydrazide, ring-clos- ing macrolactam formation by the hydrazide-based ligation, and final oxidative folding to the native Cys-knot topology (Scheme 5). KB1, cO2, and MCoTI-II were selected as the representative examples for each cyclotide subfamily. The linear peptide hydrazides were readily prepared through standard Fmoc SPPS from the hydrazine-Trt(2-Cl) resin. The isolated yields of the linear peptide hydrazides (based on the initial resin loading) were 28 %, 36 % and 30 % for KB1 (29 mer), cO2 (31 mer), and MCoTI-II (34 mer), respectively. Ring-closing liga- tions of the peptide hydrazides were then conducted by using the optimized protocol shown in Table 1. HPLC monitoring of the ligation processes indicated that the cyclization can be completed within two hours. The HPLC yields were 93, 91, and 91% (with 54, 61, and 50% isolated yields) based on the linear peptides for KB1, cO2, and MCoTI-II, respectively (Figure 2). Finally, the folding of the three cyclotides was conducted by using the previously established protocols (i.e. air oxida- tion in the presence of glutathione). The MALDI-TOF masses and HPLC retention time of the folded proteins were fully consistent with the previous studies.
In summary, we have introduced a new method for lactamization of fully unprotected peptides in an epimerization-free manner. This method is developed on the basis of the ligation of peptide hydrazides. Its major advantage is that peptide hydrazides can be easily prepared by using the routine use of Fmoc solid-phase peptide synthesis. Thus the present method can be used to synthesize various cyclic peptides and cyclic proteins with increased simplicity and lower costs. The applica- tion of the method is demonstrated by the efficient synthesis of KB1, cO2, and MCoTI-II, which belong to three subfamilies of cyclotides.
Acknowledgements
This study was supported by NSFC (No. 20932006, 91013007) and the national “973” grants from the Ministry of Science and Tech- nology (No. 2011CB965300).
- a) Y. Hamada, T. Shioiri, Chem. Rev. 2005, 105, 4441; b) A. R. Horswill, S. J. Benkovic, Cell Cycle 2005, 4, 552; c) D. J. Craik, Science 2006, 311, 1563 – 1564 ; d) C. J. White, A. K. Yudin, Nature Chem. 2011, 3, 509 ; e) D. J. Craik, N. L. Daly, Curr. Opin. Chem. Biol. 2011, 15, 362; f) R. M. J. Liskamp, D. T. S. Rijkers, J. A. W. Kruijtzer, J. Kemmink, ChemBioChem 2011, 12, 1626.
- a) J. N. Lambert, J. P. Mitchell, K. D. Roberts, J. Chem. Soc. Perkin Trans. 1 2001, 471; b) M. C. Alcaro, G. Sabatino, J. Uziel, M. Chelli, M. Ginanne- schi, P. Rovero, A. M. Papini, J. Pept. Sci. 2004, 10, 218; c) A. M. Felix, C. T. Wang, E. P. Heimer, A. J. Fournier, Int. J. Pept. Protein Res. 1988, 31, 231; d)F. Albericio, R.P. Hammer, C. Garciaecheverria, M.A. Molins, J.L. Chang, M. C. Munson, M. Pons, E. Giralt, G. Barany, Int. J. Pept. Protein Res. 1991, 37, 402; e) G. Barany, J. Tulla-Puche, J. Org. Chem. 2004, 69, 4101.
- a) U. Schmidt, J. Langner, J. Pept. Res. 1997, 49, 67; b) L. S. Zhang, J. P. Tam, J. Am. Chem. Soc. 1999, 121, 3311.
- a) L. S. Zhang, J. P. Tam, J. Am. Chem. Soc. 1997, 119, 2363; b) J. P. Tam, Y. A. Lu, Q. T. Yu, J. Am. Chem. Soc. 1999, 121, 4316; c) J. P. Tam, Y. A. Lu, J. L. Yang, K. W. Chiu, Proc. Natl. Acad. Sci. USA 1999, 96, 8913.
- a) J. A. Camarero, T. W. Muir, Chem. Commun. 1997, 1369; b) J. A. Camar- ero, J. Pavel, T. W. Muir, Angew. Chem. 1998, 110, 361; Angew. Chem. Int. Ed. 1998, 37, 347; c) E. A. George, R. P. Novick, T. W. Muir, J. Am. Chem. Soc. 2008, 130, 4914.
- a) Y. Shao, W. Y. Lu, S. B. H. Kent, Tetrahedron Lett. 1998, 39, 3911; b) L. Z. Yan, P. E. Dawson, J. Am. Chem. Soc. 2001, 123, 526; c) D. Hilvert, R. Qua- derer, Chem. Commun. 2002, 2620; d) V. M. F. Cardona, O. Hartley, P. Botti, J. Pept. Res. 2003, 61, 152; e) C. T. Pool, J. G. Boyd, J. P. Tam, J. Pept. Res. 2004, 63, 223; f) J. A. Camarero, B. J. Hackel, J. J. de Yoreo, A. R. Mitchell, J. Org. Chem. 2004, 69, 4145; g) D. J. Craik, R. J. Clark, Bio- polymers 2010, 94, 414; h) N. Umezawa, Y. Noro, K. Ukai, N. Kato, T. Hi- guchi, ChemBioChem 2011, 12, 1694; i) D. Macmillan, M. De Cecco, N. L. Reynolds, L. F. A. Santos, P. E. Barran, J. R. Dorin, ChemBioChem 2011, 12, 2133.
- Reviews: a) P. E. Dawson, S. B. H. Kent, Annu. Rev. Biochem. 2000, 69, 923; b) C. P. R. Hackenberger, D. Schwarzer, Angew. Chem. Int. Ed. 2008, 47, 10030 ; c) S. B. H. Kent, Chem. Soc. Rev. 2009, 38, 338.
- P. Botti, T. D. Pallin, J. P. Tam, J. Am. Chem. Soc. 1996, 118, 10018.
- R. Kleineweischede, C. P. R. Hackenberger, Angew. Chem. 2008, 120, 6073; Angew. Chem. Int. Ed. 2008, 47, 5984.
- Ahsanullah, J. Rademann, Angew. Chem. 2010, 122, 5506; Angew. Chem. Int. Ed. 2010, 49, 5378.
- a) D. Crich, A. Banerjee, J. Am. Chem. Soc. 2007, 129, 10064; b) C. Haase, H. Rohde, O. Seitz, Angew. Chem. 2008, 120, 6912; Angew. Chem. Int. Ed. 2008, 47, 6807; c) J. Chen, Q. Wan, Y. Yuan, J. Zhu, S. J. Danishefsky, Angew. Chem. 2008, 120, 8649; Angew. Chem. Int. Ed. 2008, 47, 8521; d) R. L. Yang, K. K. Pasunooti, F. P. Li, X. W. Liu, C. F. Liu, J. Am. Chem. Soc. 2009, 131, 13592; e) K. S. A. Kumar, A. Brik, J. Pept. Sci. 2010, 16, 524; f) Z. Harpaz, P. Siman, K. S. A. Kumar, A. Brik, ChemBioChem 2010, 11, 1232 ; g) H. Rohde, O. Seitz, Biopolymers 2010, 94, 551; h) S. Shang, Z. Tan, S. Dong, S. J. Danishefsky, J. Am. Chem. Soc. 2011, 133, 10784.
- G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui, L. Liu, Angew. Chem. Int. Ed. 2011, 50, 7645.
- a) M. Schnçlzer, P. Alewood, A. Jones, D. Alewood, S. B. H. Kent, Int. J. Pept. Res. Ther. 2007, 13, 31.
- a) B. J. Backes, A. A. Virgilio, J. A. Ellman, J. Am. Chem. Soc. 1996, 118, 3055; b)R. Ingenito, E. Bianchi, D. Fattori, A. Pessi, J. Am. Chem. Soc. 1999, 121, 11369 ; c) M. Schaffrath, C. Unverzagt, Angew. Chem. 2005, 117, 1677; Angew. Chem. Int. Ed. 2005, 44, 1650; d)F. Mende, O. Seitz, Angew. Chem. 2007, 119, 4661; Angew. Chem. Int. Ed. 2007, 46, 4577; e)F. Mende, M. Beisswenger, O. Seitz, J. Am. Chem. Soc. 2010, 132, 11110.
- a) J. B. Blanco-Canosa, P. E. Dawson, Angew. Chem. 2008, 120, 6957; Angew. Chem. Int. Ed. 2008, 47, 6851; b) A. P. Tofteng, K. K. Sorensen, K. W. Conde-Frieboes, T. Hoeg-Jensen, K. J. Jensen, Angew. Chem. 2009, 121, 7547; Angew. Chem. Int. Ed. 2009, 48, 7411.
- H. E. Hallen, H. Luo, J. S. Scott-Craig, J. D. Walton, Proc. Natl. Acad. Sci. USA 2007, 104, 19097.
- G. Stavropoulos, D. Gatos, V. Magafa, K. Barlos, Lett. Pept. Sci. 1995, 2, 315.
- E. C. B. Johnson, S. B. H. Kent, J. Am. Chem. Soc. 2006, 128, 6640.
- N.-H. Tan, J. Zhou, Chem. Rev. 2006, 106, 840.
- Q. Wan, S. J. Danishefsky, Angew. Chem. 2007, 119, 9408; Angew. Chem. Int. Ed. 2007, 46, 9248.
- B. L. Pentelute, S. B. H. Kent, Org. Lett. 2007, 9, 687.
- J. Chen, J. D. Warren, B. Wu, G. Chen, Q. Wan, S. J. Danishefsky, Tetrahe- dron Lett. 2006, 47, 1969.
- a) D. J. Craik, N. L. Daly, T. Bond, C. Waine, J. Mol. Biol. 1999, 294, 1327; b) D. J. Craik, N. L. Daly, J. Mulvenna, M. R. Plan, M. Trabi, Curr. Protein Pept. Sci. 2004, 5, 297; c) D. J. Craik, A. C. Conibear, J. Org. Chem. 2011, 76, 4805.
- a) M. L. Colgrave, D. J. Craik, Biochemistry 2004, 43, 5965; b) K. R. Gustaf- son, T. C. McKee, H. R. Bokesch, Curr. Protein Pept. Sci. 2004, 5, 331; c) U. Goransson, E. Svangard, P. Claeson, L. Bohlin, Curr. Protein Pept. Sci. 2004, 5, 317; d) N. L. Daly, D. J. Craik, Future Med. Chem. 2009, 1, 1613; e) S. T. Henriques, Drug Discovery Today 2010, 15, 57; f) N. L. Daly, D. J. Craik, Curr. Opin. Chem. Biol. 2011, 15, 362.
- a) J. P. Tam, Y. A. Lu, Tetrahedron Lett. 1997, 38, 5599; b) N. L. Daly, S. Love, P. F. Alewood, D. J. Craik, Biochemistry 1999, 38, 10606; c) P. Thon- gyoo, E. W. Tate, R. J. Leatherbarrow, Chem. Commun. 2006, 2848; d) U. Goransson, T. L. Aboye, R. J. Clark, D. J. Craik, ChemBioChem 2008, 9, 103; e) S. Park, S. Gunasekera, T. L. Aboye, U. Goransson, Int. J. Pept. Res. Ther. 2010, 16, 167; f) J.-S. Zheng, H.-N. Chang, J. Shi, L. Liu, Sci. China Chem. 2012, 55, 64.
- U. Goransson, T. L. Aboye, R. J. Clark, R. Burman, M. B. Roig, D. J. Craik, Antioxid. Redox Signaling 2011, 14, 77.